I've not been blogging for a while, due to a pretty time-consuming combination of writing papers, making presentations and writing my thesis (on-going!).
Anyway, a quick post in the vein of it-could-have-saved-me-a-minute-if-I'd-seen-it: what to do when you've made a PDF in LaTeX which includes PDF figures make in Inkscape, but the text isn't appearing?
If you've checked out the two obvious search hits and still no text, try unticking both the 'Convert text to paths' and 'PDF+LaTeX...' options (the two that you'd think should ensure your text is correctly displayed!), it worked for me.
Wednesday, 22 October 2014
Wednesday, 8 October 2014
Happy Pearl anniversary PCR
Polymerase chain reaction, arguably the single most important molecular biological technique (or so I, with my training in molecular genetics might claim) turns 30 this year. The importance of this technique, that allows for easy, cheap and reliable amplification of a small amount of DNA into a large amount, cannot be understated.
On a personal level, every single project I've ever worked on in my seven or eight years in the lab has used PCR in one flavour or another, which in total probably accounts for more than half of the experiments I've ever done. PCR is a big deal to me as well the world.
So when Kary Mullis, the scientist who invented - and later won the Nobel prize for inventing - PCR came to London this week to give a talk commemorating this anniversary I jumped at the chance. (As an added bonus it was hosted in the Royal Institution, and I love a good poke around old scientific societies).
It was a nice talk. In all honesty, I went in with the expectation that he might air one of his less informed (or less palatable) personal views, but I was pleasantly surprised; the talk was pretty much entirely about the story of the making of PCR.
And a good talk it was. Kary is an engaging, funny, charismatic man. The story goes that before PCR he was a chemist arduously making oligonucleotides to order, saying that they "could make maybe three 15-base long oligonucleotides in a month". To someone who grew up being able to order a multitude of oligos of up to 100 odd bases, with all sorts of fancy modifications, and get them all delivered in a week or two, this dates the story pretty well.
After a friend and colleague figured out a way to automate the synthesis reaction, Mullis needed to think up something to do with all these oligos and keep himself and his labmates in work. Then, on that famous winding drive home, he had his Eureka moment.
It was an endearing talk, and I would recommend people listen to it (if they're able), I'm just sadly not sure how much of it I believe. It was all a bit too practised, a bit too perfect ("and then I turned to my girlfriend of the time and said, 'you know, if this works I might just get the Nobel prize"), and falling a little too much into "these are all the journals that turned me down, look at me now" category.
He even specifically went to great lengths to say how completely novel it was and that nothing even close had ever been done before, which I knew for a fact was just plain not true. Now obviously it would have been entirely possible for him not to have heard about this similar, pre-existing (but inferior) technique at the time, but to be saying in no uncertain terms that PCR was entirely novel 30 years down the line seems a bit rich. I'm not trying to discredit or impugn here; as I said, the development of PCR was a genuine feat of human ingenuity that boosted the biological sciences years forwards, but come on Kary, you got the Nobel, no need to be stingy with credit (or facts).
Unfortunately it was immediately followed by a rather dry panel section with a few pharma top bods talking about how to solve the problem of antibiotic resistance (as Mullis' current venture is developing drugs to help tackle said problem). The topic is undeniably incredibly important, but the dialogue was frankly a little too economic for my tastes: I can see how finding out just how much we need to grease industry to get them to make the drugs we need is important, it's just not how I'd choose to spend my evenings when I'm crammed into tiny seminar seats. That said, one of the speakers was Andrew Dillon, the CEO of NICE, who seemed to be a wonderfully sensible man (and thankfully bereft of the horrible corporate-talk that plagued some of the other speakers), which put a pleasant light on the end of the evening.
Too long without writing a blog post, and I'm rambling. I only really meant to write one thing, and that is happy Pearl anniversary PCR, keep up the good work.
On a personal level, every single project I've ever worked on in my seven or eight years in the lab has used PCR in one flavour or another, which in total probably accounts for more than half of the experiments I've ever done. PCR is a big deal to me as well the world.
So when Kary Mullis, the scientist who invented - and later won the Nobel prize for inventing - PCR came to London this week to give a talk commemorating this anniversary I jumped at the chance. (As an added bonus it was hosted in the Royal Institution, and I love a good poke around old scientific societies).
It was a nice talk. In all honesty, I went in with the expectation that he might air one of his less informed (or less palatable) personal views, but I was pleasantly surprised; the talk was pretty much entirely about the story of the making of PCR.
And a good talk it was. Kary is an engaging, funny, charismatic man. The story goes that before PCR he was a chemist arduously making oligonucleotides to order, saying that they "could make maybe three 15-base long oligonucleotides in a month". To someone who grew up being able to order a multitude of oligos of up to 100 odd bases, with all sorts of fancy modifications, and get them all delivered in a week or two, this dates the story pretty well.
After a friend and colleague figured out a way to automate the synthesis reaction, Mullis needed to think up something to do with all these oligos and keep himself and his labmates in work. Then, on that famous winding drive home, he had his Eureka moment.
It was an endearing talk, and I would recommend people listen to it (if they're able), I'm just sadly not sure how much of it I believe. It was all a bit too practised, a bit too perfect ("and then I turned to my girlfriend of the time and said, 'you know, if this works I might just get the Nobel prize"), and falling a little too much into "these are all the journals that turned me down, look at me now" category.
He even specifically went to great lengths to say how completely novel it was and that nothing even close had ever been done before, which I knew for a fact was just plain not true. Now obviously it would have been entirely possible for him not to have heard about this similar, pre-existing (but inferior) technique at the time, but to be saying in no uncertain terms that PCR was entirely novel 30 years down the line seems a bit rich. I'm not trying to discredit or impugn here; as I said, the development of PCR was a genuine feat of human ingenuity that boosted the biological sciences years forwards, but come on Kary, you got the Nobel, no need to be stingy with credit (or facts).
Unfortunately it was immediately followed by a rather dry panel section with a few pharma top bods talking about how to solve the problem of antibiotic resistance (as Mullis' current venture is developing drugs to help tackle said problem). The topic is undeniably incredibly important, but the dialogue was frankly a little too economic for my tastes: I can see how finding out just how much we need to grease industry to get them to make the drugs we need is important, it's just not how I'd choose to spend my evenings when I'm crammed into tiny seminar seats. That said, one of the speakers was Andrew Dillon, the CEO of NICE, who seemed to be a wonderfully sensible man (and thankfully bereft of the horrible corporate-talk that plagued some of the other speakers), which put a pleasant light on the end of the evening.
Too long without writing a blog post, and I'm rambling. I only really meant to write one thing, and that is happy Pearl anniversary PCR, keep up the good work.
Thursday, 4 September 2014
Recovering TCR sequences from ENCODE RNASeq data (or not)
You might have seen that there's recently been another wave of papers and data from the ENCODE project.
Browsing through their data, I noticed one sample was a large RNASeq experiment using peripheral blood mononuclear cells (PBMC) as the input cell type: as this will contain a lot of T cell RNA, I thought I'd have a look for some TCRs!
It was a pretty fruitless endeavour. The smaller of the two fastqs, clocking in with 27.5 million reads, yielded some 70-odd TCRs all told. The data, being 100bp reads from presumably randomly sheared/reverse transcribed just doesn't catch enough V(D)J windows to consistently mine (without any assembly).
The larger file was still running the next day having only spat out another 70, so I called it quits. Rich data sets they may be, but appropriate for Decombinator they aint.
Oh well, back to the drawing board.
Browsing through their data, I noticed one sample was a large RNASeq experiment using peripheral blood mononuclear cells (PBMC) as the input cell type: as this will contain a lot of T cell RNA, I thought I'd have a look for some TCRs!
It was a pretty fruitless endeavour. The smaller of the two fastqs, clocking in with 27.5 million reads, yielded some 70-odd TCRs all told. The data, being 100bp reads from presumably randomly sheared/reverse transcribed just doesn't catch enough V(D)J windows to consistently mine (without any assembly).
The larger file was still running the next day having only spat out another 70, so I called it quits. Rich data sets they may be, but appropriate for Decombinator they aint.
Oh well, back to the drawing board.
Saturday, 26 July 2014
See an error in a database? Let someone know!
Anyone who does
any T cell receptor analysis will know IMGT (the ImmunoGenetics
DataBase), the repository for all things TCR and Ig. You either use
it, or you're one of those annoying people that makes me have to drag
up all the tables of outdated nomenclatures.
Much like any
resource, IMGT has it good points (simple and highly useful features
like GENE-DB and LocusView in particular) and its bad (the less said
about LIGM-DB the better).
However, again
like any resource, it's only as good as the data stored in it. The
data in it, as far as I can tell, is pretty damn good (and I use it a
lot). I guess that's why
they got to be in charge of all the data in the first place.
As such, when I
recently found an error in a sequence*, I made sure to let them know:
I certainly get a lot of mileage out of their data, it's only fair
that I pay them back (and pay it forward to others) by ensuring the
data that is there is good.
It's always a
little nerve-inducing, being a PhD student emailing senior doctors
and professors to let them know of a mistake you've discovered, but
as hoped the information was very warmly received, and I'm told that the
error will be corrected.
Science has to be
self-correcting to stop errors lingering and spreading; firing a
quick email off to correct an annotation might not seem like much,
but if it stops one person going through the same short time of
confusion that you went through unravelling the mistake then you've
done a net service to the world.
* For the people that found their way here suffering from this particular error, here's what I found. I was looking at
the TCR leader regions(the mono-spliced section of the transcript
between the start of translation and the beginning of the V region
which encodes the localisation signal peptide), when I noticed that
one gene never seemed to produce functional transcripts. It turned
out that while some of the entries for the human alpha gene
TRAV29,DV5 were correct, if you downloaded the L-PART2 region alone
the sequence produced actually contains a section of the start of
the V gene. So, instead of reading 'GGGTAAAC', it reads
'GGGTAAACAGTCAACAGAAGAATGAT'. I just checked and it still
gives the old sequence, but I assume there's a lag time for databases
to update.
Tuesday, 3 June 2014
The Inner Army Crept Up On Me
Tonight saw my
maiden voyage into the world of giving public engagement talks about
science. It came as a particular surprise because I thought I was
just the delivery boy.
The event was The
Inner Army, an hour of immunological discussion at the CheltenhamScience Festival, with Professors Susan
Lea and Clare
Bryant presenting.
I'd been approached
by the British Society of
Immunology (BSI) about perhaps 3D printing some immune molecules
for the talk, after seeing some of my previous
models. I'm a big public engagement proponent, and a big fan of
the festival, having blogged
about it for my university in the first year of my PhD, so I
leapt at the chance to help out*. Plus it gave me a nice chance to
show off the demonstrative use of my models (and help justify the
time I've spent making them!).
Little did I know
that on arrival the chair for the event, the illustrious Vivienne
Parry (who was originally an immunologist herself) decided to get
hold of an extra chair and mic and throw me up on stage as well!
It was – I think –
a fun and informative event. However, I can take no credit for any of
it (except for most of the models): I choked! Give me small numbers
of people and I'll happily ramble on about adaptive immunity to the
cows come home. Sit me down next to two prominent professors in front
of ninety people and ask me to talk about structural innate immunity
and it turns out I get a bit tongue-tied. Live and learn!
I was very happy to
see how involved the audience seemed to be with the models
(particularly the first row, which seemed to be largely composed of
BSI and British
Crystallographic Association (BCA) members), which was very
gratifying. It was also lovely to see the general public engaging
with immunology in person, which isn't something I get to see on a
daily basis.
For the moment I'd
be lying if I said I wasn't more comfortable on the other side of the
spotlights blogging about the event (which I suppose is what I'm
doing now). This isn't something that comes naturally to me, or (I
suspect) a lot of science post-graduates; it just isn't a skill we
get to practise much in our day-to-day workings.
However, engaging
with the public remains an important task for scientists, both to
justify the tax-payer money we spend and to share the love of
uncovering the secrets of the universe with fellow curious minds, so
I shall definitely try again. Next time though, I plan to stick to
TCRs.
* NB I plan to share photos of the models I made for the speakers in a
future post, but as the models dispersed to the relevant speakers after the talk I have to dig them
Sunday, 4 May 2014
Translating TCR sequences addendum: not as easy as FGXG
I recently wrote a blog post about the strategies used to translate T-cell receptor nucleotides en masse and extract (what can arguable be considered) the useful bit: the CDR3.
In that talk I touched on the IMGT-definition of the CDR3: it runs from the second conserved cysteine in the V region to the conserved FGXG motif in the J. Nice and easy, but we have to remember that it's the conserved bit that's key here: there are other cysteines to factor in, and there are a few germline J genes that don't use the typical FGXG motif.
However even that paints too simple a picture, so here's a quick follow up point:
These are human-imposed definitions, based more on convenience for human-understanding than biological necessity. The fact is that we might well produce a number of TCRs that don't make use of these motifs at all, but that are still able to function perfectly well; assuming the C/FGXG motifs have function, it's possible alternative motifs might compensate for these.
I have examples in my own sequence data that appear to clearly show these motifs having been deleted into, and then replaced with different nucleotides encoding the same. Alternative residues must certainly be introduced on occasion, and I'd be surprised if none of these make it through selection; we just don't see these because we aren't able to generate rules to computationally look for these.
I actually even recently found such an example with verified biological activity: this paper sequenced tetramer-sorted HIV-reactive T-cells, revealing one that contained an alpha chain using the CDR3 'CAVNIGFGNVLHCGSG'.*
For the majority of analyses, looking for rare exceptions to rules probably won't make much difference. However as we increase the resolution and throughput of our experiments, we're going to find more and more examples of things which don't fit the tidy rules we made up when we weren't looking so deeply. If we're going to get the most out of our 'big data', we need to be ready for them
* I was looking through the literature harvesting CDR3s, which reminds me of another point I want to make. Can I just ask, from the bottom of my heart, for people to put their CDR3s in sensible formats so that others can make use of them? Ideally, give me the nucleotide sequence. Bare minimum, give me the CDR3 sequence as well as which V and J were used (and while I stick to IMGT standards, I won't judge you if you don't - but do say which standards you are using!). Most of all, and I can't stress this enough, please please PLEASE make all DNA/amino acid sequences copyable.**
** Although spending valuable time copying out or removing PDF-copying errors from hundreds of sequences drives me ever so slightly nearer to a breakdown, it does allow me to play that excellent game of "what's the longest actual word I can find in biological sequences". For CDR3s, I'm up to a sixer with 'CASSIS'.
In that talk I touched on the IMGT-definition of the CDR3: it runs from the second conserved cysteine in the V region to the conserved FGXG motif in the J. Nice and easy, but we have to remember that it's the conserved bit that's key here: there are other cysteines to factor in, and there are a few germline J genes that don't use the typical FGXG motif.
However even that paints too simple a picture, so here's a quick follow up point:
These are human-imposed definitions, based more on convenience for human-understanding than biological necessity. The fact is that we might well produce a number of TCRs that don't make use of these motifs at all, but that are still able to function perfectly well; assuming the C/FGXG motifs have function, it's possible alternative motifs might compensate for these.
I have examples in my own sequence data that appear to clearly show these motifs having been deleted into, and then replaced with different nucleotides encoding the same. Alternative residues must certainly be introduced on occasion, and I'd be surprised if none of these make it through selection; we just don't see these because we aren't able to generate rules to computationally look for these.
I actually even recently found such an example with verified biological activity: this paper sequenced tetramer-sorted HIV-reactive T-cells, revealing one that contained an alpha chain using the CDR3 'CAVNIGFGNVLHCGSG'.*
For the majority of analyses, looking for rare exceptions to rules probably won't make much difference. However as we increase the resolution and throughput of our experiments, we're going to find more and more examples of things which don't fit the tidy rules we made up when we weren't looking so deeply. If we're going to get the most out of our 'big data', we need to be ready for them
* I was looking through the literature harvesting CDR3s, which reminds me of another point I want to make. Can I just ask, from the bottom of my heart, for people to put their CDR3s in sensible formats so that others can make use of them? Ideally, give me the nucleotide sequence. Bare minimum, give me the CDR3 sequence as well as which V and J were used (and while I stick to IMGT standards, I won't judge you if you don't - but do say which standards you are using!). Most of all, and I can't stress this enough, please please PLEASE make all DNA/amino acid sequences copyable.**
** Although spending valuable time copying out or removing PDF-copying errors from hundreds of sequences drives me ever so slightly nearer to a breakdown, it does allow me to play that excellent game of "what's the longest actual word I can find in biological sequences". For CDR3s, I'm up to a sixer with 'CASSIS'.
Wednesday, 26 March 2014
TCR trivia part one - translating T cell receptor sequences
Here's the first in what's likely to
a be pretty niche series of posts – but hopefully useful to some –
on various bits of TCR trivia I've gleaned during my PhD. This
installment: translating TCR sequences.
Whether you're doing high- or
low-throughput DNA TCR sequencing, it's highly likely that at some
point you'll want to translate your DNA into amino acid sequences.
There's usually a few main goals to
bear in mind when translating; to check the frame, and (potential)
functionality of the sequence (i.e. lacking premature stop codons),
before defining the hypervariable CDR3 region.
Assuming your reads are indel free,
sequencing variable antigen receptors will always involve a bit more
thought about checking the reading frame than regular amplicon
sequencing, due to the non-templated addition and deletion of
nucleotides that occurs during V(D)J
recombination.
So how do can we tell whether our
final recombined sequence is in frame or not?
This will mostly depend on how your
sequencing protocols work.
If, say you've somehow managed to
sequence an entire TCR mRNA sequence, then it's easy; you can just
take the sequence from the start codon of the leader region to the
stop codon of the constant region and if it divides exactly by three
then it's in frame. Assuming there's no stop codons in between and
the CDR3 checks out, chances are good your sequence encodes a
productively rearranged TCR chain.
Given today's technology, that's
unlikely to be the case; whether you're cloning into plasmids and
Sanger sequencing or throwing libraries into some next-gen machine,
chances are good that the sequence you'll be working with is actually
just a small window around the recombination site.
What's likely is that you have
amplified from a constant or J region primer at one end, to a V or
RACE/template-switching primer at the other. Sorting out the frame is
then just a matter of finding sequences on either side of the
rearrangement that you know should be in frame, and seeing whether
they are.
So far so self-explanatory. Here's
the first of the fiddlier bits of TCR trivia I want to impart, the
thing I need to remind myself of every time I tweak my TCR
translating scripts: in fully-spliced TCR message the last
nucleotide of the J region makes up the first
nucleotide of the first codon of the constant region.
This means that if you go from the
first nucleotide of the V (or the leader sequence, all functional
leader sequences being divisible by three) to either the second to
last nucleotide of the J, or the second nucleotide of the C (if
you've started with mRNA instead of gDNA) then presto, you'll be in
frame!
This feature also produces another
noteworthy feature, at least in one chain; while every TRBJ ends in
the same nucleotide, there are functional TRAJ genes ending in each
base. This means that the first residue of the constant region of the
alpha chain can be one of four different amino acids – which can be
a bit off-putting if you don't know this and you're looking at what's
supposed to be constant.
Once you've translated your TCR the
next step is to find the CDR3, which should surely be the easiest
bit; just run from the second conserved cysteine to the phenylalanine
in the FGXG motif, right, just as IMGT says? Simple, or so it might
seem.
Both sides of the CDR3 offer their
own complications. First off, finding the second conserved cysteine
isn't as easy as just picking the second C from the 5', or the
5'-most one upstream of the rearrangement, as some Vs have more than
two germline cysteines, and it's feasible that new ones might be
generated in the rearrangement. At some point you're just going to
have to do an alignment of all the different Vs, and record the
position of all the conserved cysteines.
The FGXG is also trickier than it
might seem, by merit of the fact that there are J genes in both the
alpha and gamma loci* that (while supposedly functional) lack the
motif, being either FXXG or XGXG. If you're only looking for CDR3s
matching the C to FGXG pattern, then there's going to be whole J
genes which never seem to get used by your analysis!
There you have it, a couple of little
tips that are worth bearing in mind when translating TCR sequence**.
Addendum - it's actually a little bit more complicated. Naturally.
Addendum - it's actually a little bit more complicated. Naturally.
*
If you're interested (or don't believe me), they are: TRAJ16, TRAJ33,
TRAJ38, TRGJP1 and TRGJP2.
**
Note that everything written here is based on human TCR sequence as
that's what I work with, but most of it will probably apply to other
species as well.
Sunday, 23 March 2014
TCR diversity in health and in HIV
Before I forget, here's a record of the poster I presented recently at the Quantitative Immunology workshop in Les Houches (which I blogged about the first day of last week*).
In a move I was pretty pleased with, I hosted my poster on figshare, with an additional pdf of supplementary information. (I even included a QR code on the poster linking to both, which I thought pretty cunning, but this plan was sadly scuppered by a complete lack of wifi signal.)
You can access the poster here, and the supplementary information here.
The poster gives a gives a quick glance at some of my recent work where I've been using random barcodes to error-correct deep-sequenced TCR repertoires, a technique I'm applying to comparing healthy individuals to HIV patients, both before and after three months of antiretroviral therapy.
* For those interested in a larger overview of the conference, my supervisor Benny Chain wrote a longer piece at his blog Immunology with numbers
In a move I was pretty pleased with, I hosted my poster on figshare, with an additional pdf of supplementary information. (I even included a QR code on the poster linking to both, which I thought pretty cunning, but this plan was sadly scuppered by a complete lack of wifi signal.)
You can access the poster here, and the supplementary information here.
The poster gives a gives a quick glance at some of my recent work where I've been using random barcodes to error-correct deep-sequenced TCR repertoires, a technique I'm applying to comparing healthy individuals to HIV patients, both before and after three months of antiretroviral therapy.
* For those interested in a larger overview of the conference, my supervisor Benny Chain wrote a longer piece at his blog Immunology with numbers
Monday, 10 March 2014
First day of qImmunology workshop: diversity and error
It's the first day of the
quantitative Immunology
workshop in Les Houches (#qImmLH),
and there's been a definite theme: TCR repertoire sequencing. In fact
it seems to be the main theme of the conference, with around half the
people here seemingly working on them in one sense or other – my
accommodation alone seems to be populated exclusively with us*!
Seeing as I have a bit of time today, have a quick post about it.
There's been a couple of recurrent
points which have been coming up in the talks, and are being
particularly dwelt on by the discussions from the group (being based
in a Physics retreats apparently demands that we must do as the
physicists do, and ask questions throughout a talk). Well, there have
been many but these are the two I picked up on most, but that might
just be because it's what my poster is about so I'm biased.
The first is that of error. So far,
all of our pipelines involve some amount of PCR amplification, which
adds a great deal of errors on top of whatever the sequencing
technique itself will introduce. My own supervisor Benny Chain
probably dwelt on this the longest, going over some evidence to
suggest that within a given PCR there's some variability in the
efficiency of amplification, so for lower frequency clones there's
less relationship between the number of reads coming out and the
number of original molecules of DNA that went in. However a number of
the talks touched on this, and I'm sure some of the later ones will
as well.
The second theme is that of
diversity, and how to measure it. Based on the fact all of the speakers used a different metric (and the number of questions it raised from the audience) there's clearly scope for discussion. In brief,
Encarnita
Mariotti-Ferrandiz used
a species richness index to describe the number of different unique
clonotypes in mice Treg and Teff cells, Thierry Mora used Shannon
Entropy to compare diversities of zebrafish Ig CDR3s, and Eric
Shifrut looked at Gini indexes of aging mice repertoires.
This of
course all ties in to the error of the system, as any additional
error will be likely be artificially inflating the diversity while at
the same time distorting the frequency distribution.
The
last full talk of the day ended with Aleksandra Walczak talking
through the generation of diversity in TCR repertoires, mostly just
going through the figures from the excellent Murugan
et al paper.
So
far this is shaping up to be an ideal workshop for me, what with so
many people working on and talking about the exact problems I find
myself faced with on a daily basis. That it's all taking place among
some achingly beautiful scenery is just icing on the cake.

* I know it's an awful
thing to think about, but I can't help feeling that if an avalanche
hit the resort we'd be setting the relatively young field of adaptive
repertoire sequencing back a decent way!
Wednesday, 29 January 2014
Immunological 3D printing, the how-to
Here's a quick overview of the different stages of the process I went through to make the 3D printed T-cell receptors detailed in part 1.
Part 2: the process
Now there's a couple of nice tutorials kicking around on how to 3D print your favourite protein. One option which clearly has beautiful results is to use UCSF's Chimera software, which was also the approach taken by the first protein-printing I saw. However this seemed a little full-on for my first attempts, so I opted for relatively easy approach based on PyMol, modelling software I'm already familiar with.The technique I used was covered very well in a blog post by Jessica Polka, which was then covered again in a very well written instructable. As these are both very nice resources I won't spend too long going over the same ground, but I thought it would be nice to fill in some of the gaps, maybe add a few pictures.
1: Find a protein
This should be the easy bit for most researchers (although if you work on a popular molecule finding the best crystal structure might take a little longer). Have a browse of the Protein Data Bank, download the PDB and open the molecule in PyMol.All you need to do here is hide everything, show the surface, and export as a .wrl file (by saving as VRML 2). I mean that's all you need to do. If you want to colour it in, that's totally fine too.
 |
| PyMol keeps me entertained for hours. |
2: Convert to .stl
Nice and easy; open your .wrl file in MeshLab ('Import Mesh'), and then export as a .stl, which is a very common filetype for 3D structures. |
| Say goodbye to your lovely colours. |
3: Make it printable
Now we have a format that the proprietary 3D printing software can handle. As I primarily only have access to MakerBots, I next open my stl files in MakerWare.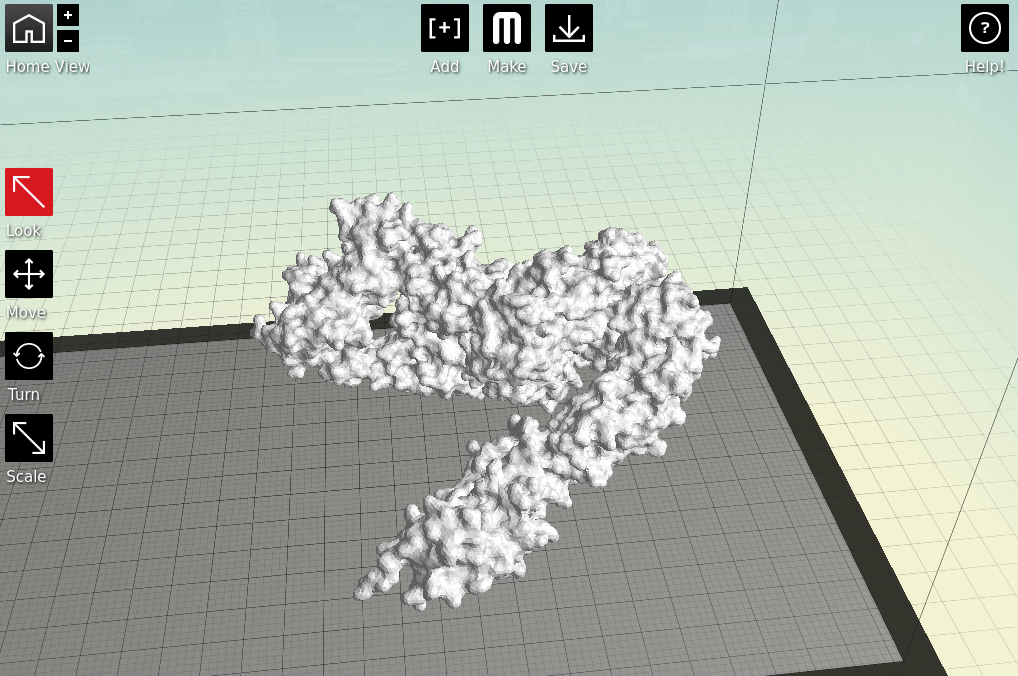 |
| Starting to feel real now, right? |
Next is the quality of the print. One factor is the scale; the bigger you make your molecule the better it will look, and likely print, at the cost of speed and plastic. Additionally you can alter the thickness of each print layer, the percentage infill (how solid the inside of the print is, up to a completely solid print) and the number of 'shells' that the print has.
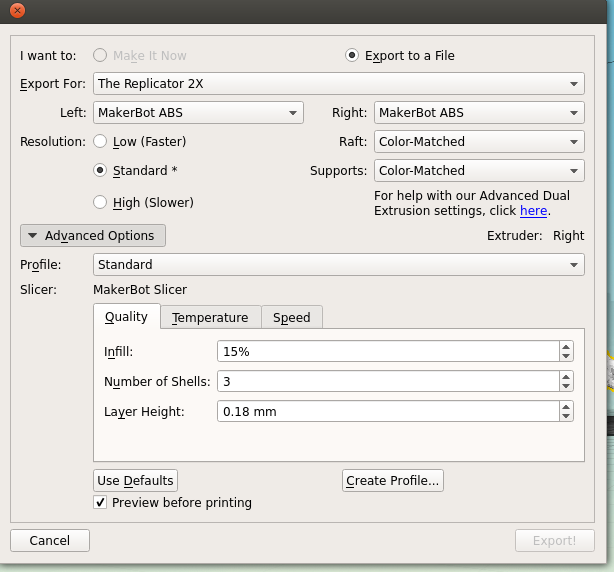 |
| Remember to tick 'Preview before printing' in order to get a time estimate. |
4: The print!
Both of my molecules so far have been printed on MakerBot Replicator 2Xs, using ABS plastic, taking between 10 and 14 hours per print due to the complexity and size of the models. This part is also nice and simple; just warm up your printer, load your file and click go. |
| A side view of the printer as it runs, detailing the raft and scaffolds that will support the various overhangs. |
 |
| The TCR begins to emerge, with hexagonal infill also visible. |
5: The tidy-up
The prints come out looking a little something like this: |
| Note the colour change where one roll of ABS ran out, and someone thoughtfully swapped it over, if sadly not for the same colour |
 |
| This green really does not photograph well. I like to pretend I was going for the nearest I could to the IMGT-approved shade of variable-region green, but really I was just picking the roll with the most ABS left to be sure it wouldn't run out again. |
 |
| My thanks go to Katharine and Mattia for de-scaffolding services rendered. |
 |
| Prepare for mess and you will not be disappointed. Instead, you will be picking tiny bits out plastic out of your hair. |
Once you've gotten all the scaffolding off, your protein should look the right shape, if a little scraggy around the edges. I've read that 3D printing people generally sometimes use fine sandpaper here to neaten up some of these edges, which I will consider in future, but generally the surface area to cover is fairly large and inaccessible, so it's not an option I've spent long dwelling on.
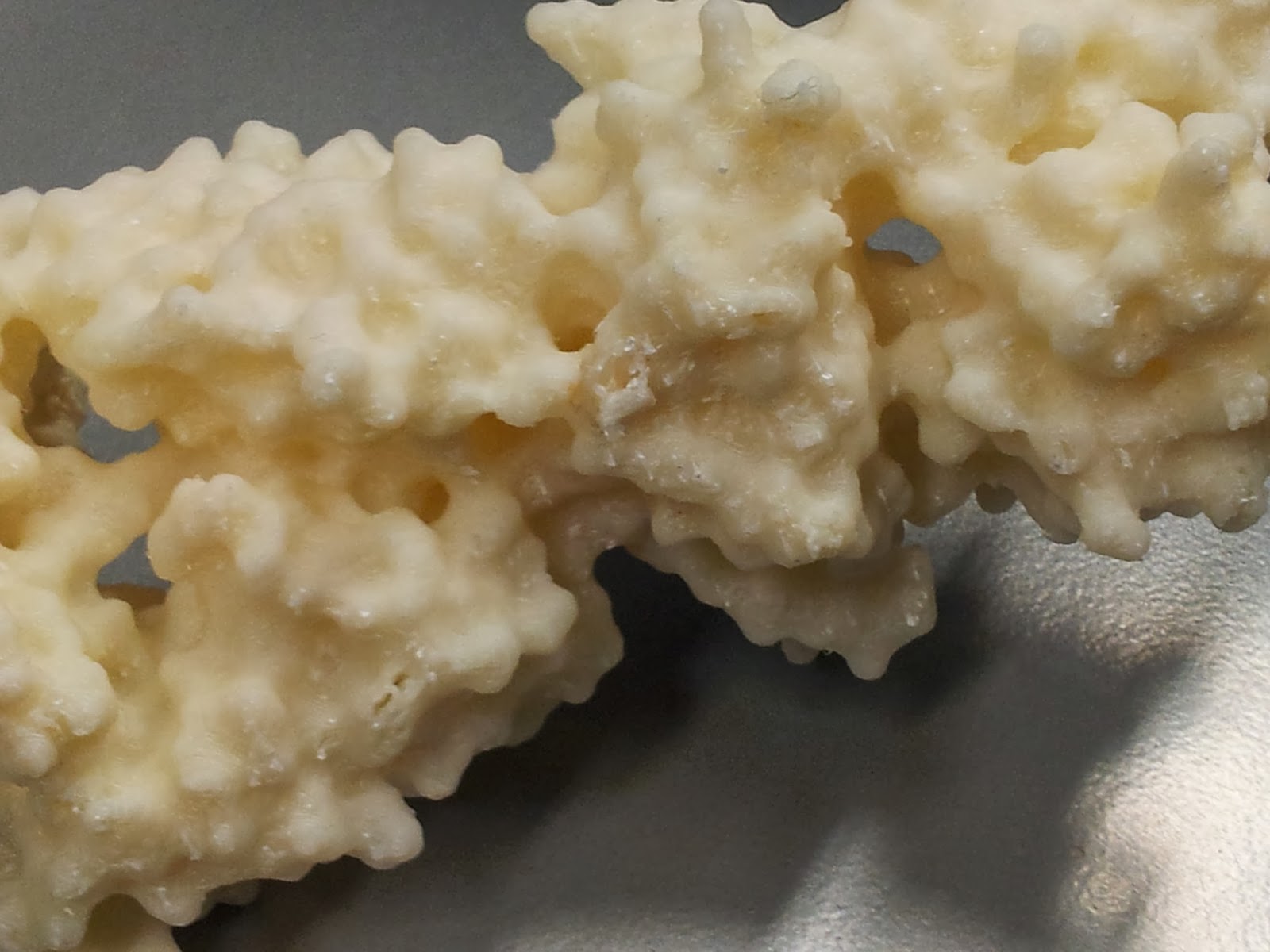 |
| The nasty underbelly of the print, after scaffold removal |
 |
| The other side is much nicer, I promise. |
 |
| Mmm, radioactive. |
However increasing the infill percentage and number of shells had one major noticeable difference; the side chains that stick out are much less fragile than they were on the first print*.
Note that this is also when the spare ABS can come in handy; dissolved in a little acetone, it readily becomes a sloppy paint, which can be slathered on to fill in any glaring flaws in the model.
I should point out that at this point the rest of the model tends to look pretty good (if I do say so myself).
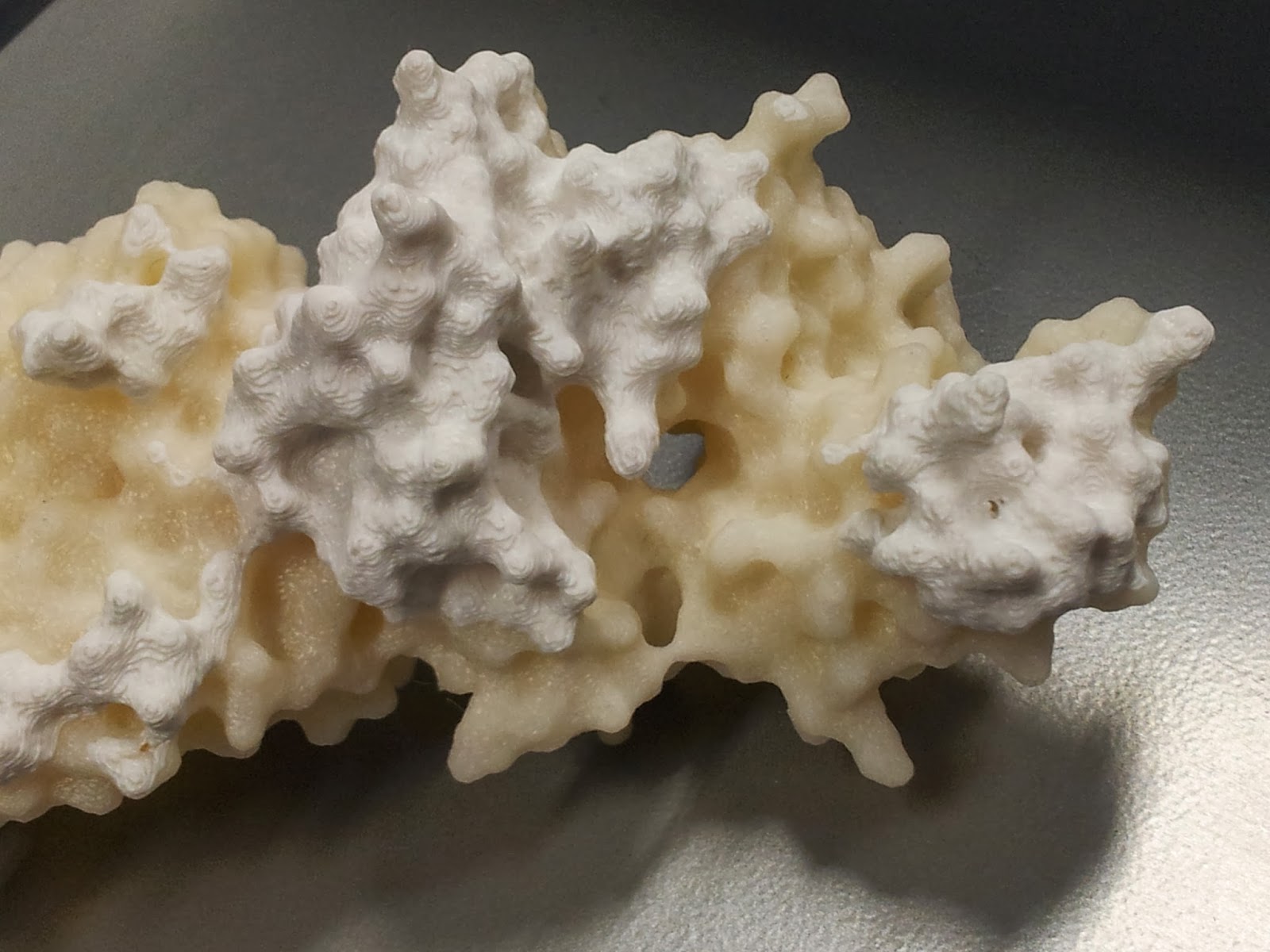 |
I got a lot of questions asking about the significance of the other colour.  |
{kind=link}
6: Smoothing
In addition to physical removal of lumps, it's also possible to smooth out the printing layers themselves by exposing the print to acetone vapour for a time, as discussed in many nice tutorials.I personally like the contour effect of the printing somewhat, and due to the delicate nature of the protrusion-heavy proteins I didn't want to go for an extreme acetone shower, but I think a light application has smoothed off some of the rougher imperfections.
This is a particularly easy thing to achieve for the average wet-lab biologist, as we have ready access to heat blocks and (usually) large beakers. Unfortunately the 3T0E, CD4 containing print was too large for any of the beakers in the lab, so I had to make do.
 |
| Nothing pleases me more than a satisfactory bodge-job. |
 |
| Still shiny - and tacky - from the acetone. |
 |
| The finished result; the model that made the immunologists of twitter all want rice pudding. What a shame the nice side had the colour change. That the acetone-smoothing appears to have affected the two colours differently suggests that different rolls of ABS do indeed have different dissolving properties. |
Or in my case, finding the time to get over there to use it.
* I tell people that I was experimenting with tactile mutational analysis, when really I just dropped the print and a couple of aromatic side chains fell off. Note that they do readily stick back on with superglue.
Immunological 3D printing
Part 1: the pictures
As a good little geek, I’ve been itching to have a play with
3D printers for a while now. At first I’d mostly contemplated what bits
and bobs I might produce for use in the lab, but then I started to see a
number of fantastic 3D printed protein models.
Form is so important to function in biology, yet a lot of
the time we biologists forget or ignore the shape and make-up of the molecules
we study. As Richard Feynman one said, “it is very easy to answer many of these
fundamental biological questions; you just look at the thing”.
3D printing protein structures represents a great opportunity
to (literally) get to grips with proteins, cells, microbes and macromolecules.
While I still recommend playing around with PDBs
to get a feel for a molecule, having an actual physical, tactile item to hold appeals to me greatly.
So when I got access to the UCL Institute of Making, I got
straight to work printing out examples of the immune molecules I study, T-cell receptors. You
can read about how I made them here. Or, if you're just here for some pretty pictures of 3D prints, continue; here are the two I've printed so far.
 |
| Here are the two finished products! I apologise for the quality: a combination of my garish fluorescent office lighting and shonky camera-phones does not a happy photo make. |
 |
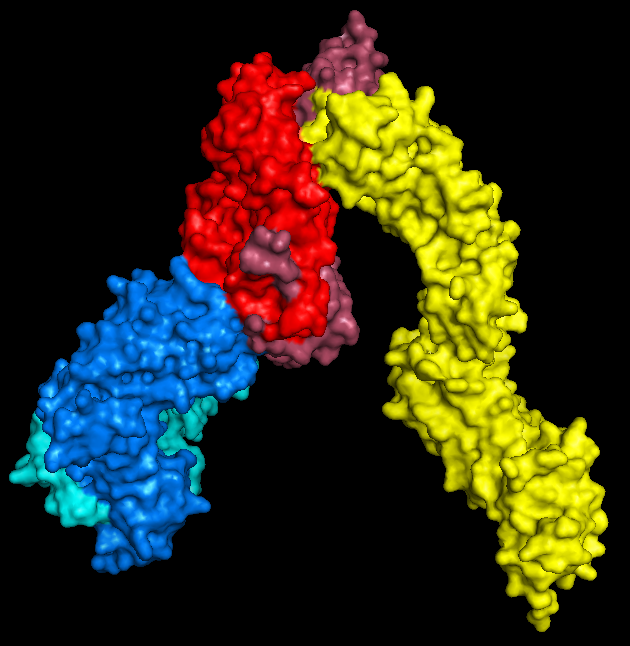
| My first try: 3WPW. This is the A6 TCR, recognising the self-peptide HuD bound in the groove of the class I HLA-A2. HLA-A2 is coloured in dark pink, with β2 microglobulin in light pink, while the alpha and beta TCR chains are shown by light and dark blue respectively. |
I particularly love the holes, crevices and caves pitted throughout the molecules. Having spent a goodly deal of time painstakingly pulling the scaffolding material out of these holes, I can confirm that you do indeed get a feel for the intricate surfaces of these structures.
You can imagine the antigen presenting cell on the left, with the T-cell on the right, as if we were watching from within the plane of the immunological synapse.
As a brief aside, in playing around with the 3PWP structure in PyMol (as detailed in an earlier blogpost) I was surprised to see the following; despite being a class I MHC (the binding grooves of which should be closed in at both ends) we can see the green of the peptide peeking out contributing to the surface mesh.
 |
| There's that HuD peptide! |
 |
| The new addition: 3T0E. This brilliant ternary structure shows another autoimmune TCR, this time recognising a class II MHC, HLA-DR4, with an additional coreceptor interaction; enter CD4! Here we have the TCR chains coloured as above, while the HLA-DR alpha and beta chains are red and magenta respectively. Leaning in to touch the (membrane-proximal) foot of the MHC is the yellow CD4. Note that I took feedback, and this time went for a colour that didn't look so rice-puddingy. |
It's also quite nice to see that despite the differences in HLA composition between classes (one MHC-encoded chain plus B2M in class I versus two MHC-encoded chains in class II), they structurally seem quite similar by eye - at least at a surface level scale.
There you have it, my first forays into 3D printing immunological molecules. Let me know what you think, if you have any ideas for future prints - I'm thinking probably some immunoglobulins for the next run - or if you're going to do any printing yourself.
Wednesday, 15 January 2014
Installing CASAVA/bcl2fastq on Ubuntu
I've been playing around with working some of my own demultiplexing scripts into my current analysis pipeline, so I thought I'd best get to grips with CASAVA, or bcl2fastq, so I can de-demultiplex (multiplex?) my MiSeq data.
The trouble is, CASAVA is not supported for my OS, Ubuntu (for reference, I'm running 13.04). Still, I thought I'd give it a try, can't take too long right? Wrong. Most painful installation ever.
I won't bore you with all the details, but here are the major stops and errors I got along the way, to help people find their way if they find themselves similarly stumped.
I started off trying to install CASAVA v1.8.2, the most up to date version I could find (before I realised that CASAVA has since turned into bcl2fastq).
I tried to build from source as per the instructions, but was unable to make:
make: *** No targets specified and no makefile found. Stop.
Checking out the log, revealed that boost was failing; it couldn't find the make file as the configure hadn't made it.
So, then I tried installing boost via apt, which let configure run fine, but everything ran aground during the make due to incompatibility issues. I should have expected this; the version on boost available through apt was several versions newer than that which comes bundled with CASAVA (1_44) (I know right, the apt version being too new? I didn't see it coming either).
Removing the version of boost I just installed seemed to now allow the bundled version to take over with the make, but now it encountered another issue:
/usr/local/CASAVA_v1.8.2/src/c++/lib/applications/AlignContig.cpp:35:32: fatal error: boost/filesystem.hpp: No such file or directory
compilation terminated.
make[2]: *** [c++/lib/applications/CMakeFiles/casava_applications.dir/AlignContig.cpp.o] Error 1
make[1]: *** [c++/lib/applications/CMakeFiles/casava_applications.dir/all] Error 2
make: *** [all] Error 2
So far so frustrated. Plan B, try and install the rpm version of the more up-to-date bcl2fastq. Now as an Ubuntu user I have no experience with rpm, but let's give it a go.
Rpm couldn't find any dependencies. I mean, any dependencies. At all. Including this:
/bin/sh is needed by bcl2fastq-1.8.4-1.x86_64
I'm not sure about a lot of the packages installed, but I know I've got sh.
OK, let's try something else; googling around the subject suggests that yum is the way to go. I tried this, it still couldn't find any of the required dependencies.
So I tried one last thing, and installed alien, and used that to install the bcl2fastq rpm...
...and it worked! Huzzah!
Although I really shouldn't have been surprised. The rpm installation even told me I should:
rpm: RPM should not be used directly install RPM packages, use Alien instead!
TL,DR: Trying to install CASAVA/bcl2fastq on Ubuntu? Try using alien to install the rpm!
Update for Ubuntu 14.04 (Trusty Tahr):
After recently updating to the newest version of Ubuntu, I've tried to follow my own advice regarding the installation of bcl2fastq, only to find that it no longer works (see below for horrendous stream of errors)!
Luckily the problem - that the newer version of Perl used in Trusty isn't compatible with bcl2fastq - had already been identified and solved by the good people over at SeqAnswers (thanks to Tony Brooks and Hiro Mishima).
"my" variable $value masks earlier declaration in same statement at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 760.
syntax error at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 747, near "$variable qw(ELAND_FASTQ_FILES_PER_PROCESS)"
Global symbol "$variable" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 749.
syntax error at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 751, near "$directory qw(ELAND_GENOME)"
Global symbol "$self" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$directory" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$project" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$sample" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$lane" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$barcode" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$reference" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
syntax error at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 761, near "}"
/usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm has too many errors.
Compilation failed in require at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment.pm line 61.
BEGIN failed--compilation aborted at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment.pm line 61.
Compilation failed in require at /usr/local/bin/configureBclToFastq.pl line 250.
BEGIN failed--compilation aborted at /usr/local/bin/configureBclToFastq.pl line 250.
The trouble is, CASAVA is not supported for my OS, Ubuntu (for reference, I'm running 13.04). Still, I thought I'd give it a try, can't take too long right? Wrong. Most painful installation ever.
I won't bore you with all the details, but here are the major stops and errors I got along the way, to help people find their way if they find themselves similarly stumped.
I started off trying to install CASAVA v1.8.2, the most up to date version I could find (before I realised that CASAVA has since turned into bcl2fastq).
I tried to build from source as per the instructions, but was unable to make:
make: *** No targets specified and no makefile found. Stop.
Checking out the log, revealed that boost was failing; it couldn't find the make file as the configure hadn't made it.
So, then I tried installing boost via apt, which let configure run fine, but everything ran aground during the make due to incompatibility issues. I should have expected this; the version on boost available through apt was several versions newer than that which comes bundled with CASAVA (1_44) (I know right, the apt version being too new? I didn't see it coming either).
Removing the version of boost I just installed seemed to now allow the bundled version to take over with the make, but now it encountered another issue:
/usr/local/CASAVA_v1.8.2/src/c++/lib/applications/AlignContig.cpp:35:32: fatal error: boost/filesystem.hpp: No such file or directory
compilation terminated.
make[2]: *** [c++/lib/applications/CMakeFiles/casava_applications.dir/AlignContig.cpp.o] Error 1
make[1]: *** [c++/lib/applications/CMakeFiles/casava_applications.dir/all] Error 2
make: *** [all] Error 2
So far so frustrated. Plan B, try and install the rpm version of the more up-to-date bcl2fastq. Now as an Ubuntu user I have no experience with rpm, but let's give it a go.
Rpm couldn't find any dependencies. I mean, any dependencies. At all. Including this:
/bin/sh is needed by bcl2fastq-1.8.4-1.x86_64
I'm not sure about a lot of the packages installed, but I know I've got sh.
OK, let's try something else; googling around the subject suggests that yum is the way to go. I tried this, it still couldn't find any of the required dependencies.
So I tried one last thing, and installed alien, and used that to install the bcl2fastq rpm...
alien -i bcl2fastq-1.8.4-Linux-x86_64.rpm...and it worked! Huzzah!
Although I really shouldn't have been surprised. The rpm installation even told me I should:
rpm: RPM should not be used directly install RPM packages, use Alien instead!
TL,DR: Trying to install CASAVA/bcl2fastq on Ubuntu? Try using alien to install the rpm!
Update for Ubuntu 14.04 (Trusty Tahr):
After recently updating to the newest version of Ubuntu, I've tried to follow my own advice regarding the installation of bcl2fastq, only to find that it no longer works (see below for horrendous stream of errors)!
Luckily the problem - that the newer version of Perl used in Trusty isn't compatible with bcl2fastq - had already been identified and solved by the good people over at SeqAnswers (thanks to Tony Brooks and Hiro Mishima).
"my" variable $value masks earlier declaration in same statement at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 760.
syntax error at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 747, near "$variable qw(ELAND_FASTQ_FILES_PER_PROCESS)"
Global symbol "$variable" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 749.
syntax error at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 751, near "$directory qw(ELAND_GENOME)"
Global symbol "$self" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$directory" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$project" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$sample" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$lane" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$barcode" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
Global symbol "$reference" requires explicit package name at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 753.
syntax error at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm line 761, near "}"
/usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment/Config.pm has too many errors.
Compilation failed in require at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment.pm line 61.
BEGIN failed--compilation aborted at /usr/local/lib/bcl2fastq-1.8.4/perl/Casava/Alignment.pm line 61.
Compilation failed in require at /usr/local/bin/configureBclToFastq.pl line 250.
BEGIN failed--compilation aborted at /usr/local/bin/configureBclToFastq.pl line 250.
Subscribe to:
Posts (Atom)